Complex biophysical processes such as protein folding and molecular recognition underlie the function of all biological systems. Although the function of these systems is generally understood, insights into the microscopic origins of this behavior are still lacking. The challenge of understanding and predicting the macroscopic behavior of complex biological systems on the basis of the atomic and molecular level interactions is what motivates our research.Studies in our group aim to explore the links between protein sequence, structure, conformational dynamics and molecular recognition to identify the interactions responsible for defined biological (mis)functions. These objectives are pursued through a combination of existing and newly developed computational and theoretical techniques. Insights gained from these studies are used to define new principles for rational protein-design, drug-design and drug-screening. The strength of our research relies on the integration of new computational methodologies with collaborative experimental approaches to provide valuable mechanistic interpretations and validations at the molecular level of the theoretical predictions.Our research work is thus undertaken in the areas of protein structure modeling and dynamics, structural biology and proteomics, ligand-protein docking, drug design and screening, and chemical genomics.
To target this complexity from an experimental point of view and translate findings into new reagents and technologies, our lab is strongly integrated with the Microanalytics and Peptide and Protein Chemistry Lab in the new ICRM-BEAM unit.
Research Projects
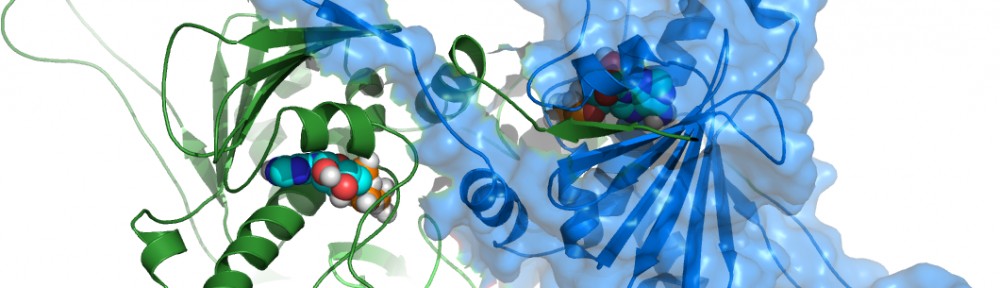
1. Computational Structural Pharmacogenomics-Dynamics Based Drug Design. Integrating molecular dynamics, computational physical-chemistry and bioinformatics approaches to study the dynamics and specificity of protein-protein, peptide-protein and protein-small molecule binding: translation of the information into pharmacophore models for drug design and screening is used to rationally design new molecules taking the full flexibility of the receptor and the ligand into account. Applications include the identification of new leads for the development of targeted cancer therapeutics for the inhibition of Hsp90 and FGF-2.
In this context, a significant effort in our research is devoted to the development of new computational/theoretical tools for the characterization of allosteric mechanisms in proteins and for the ab-initio prediction of possible allosteric binding sites. The broad objective is to identify the molecular effects of sequence-structure perturbation that are responsible for the modulation of the activity and recognition properties in proteins. These concepts are translated into new methods for the discovery of small molecule therapeutics for cancer.
2. Chaperone-Mediated Protein Folding Mechanisms. Protein Folding and Misfolding. Computer simulations and theoretical approaches to understand the mechanisms of coupling between binding and protein dynamics in chaperone mediated pathways. This entails mainly the analysis of local and global dynamics of Hsp90, analysis of Hsp90-protein recognition and binding. Despite being based on the specific case of Hsp90, the methods are fully general and are being applied to a variety of protein systems.
In the folding field, we are active in the development of atomic-resolution physics based models for the all-atom prediction of protein structure and (mis)folding mechanisms from sequence information. The models shed light on the effects of (genetic) mutations on the structural and aggregation properties of sequences.
3. Prediction of the Structural Determinants of Protein-Antibody and Protein-Protein Interactions. Current data accumulating from reverse vaccinology studies show that only a small fraction of surface-exposed proteins appears to elicit antibodies with bactericidal activity. By using information generated by reverse vaccinology projects we are developing and applying a novel integrated approach to single out the structural requirements for viable bactericidal vaccine candidates, and will develop bioinformatics tools to predict compliance with such structural requirements. To this end, a systematic analysis of sequence, structure, dynamics and interactions of selected protein targets is undertaken. New protein-interaction prediction methods based only on structural and energetic information have been developed for the particular case of Protein-Antibody recognition. The methods are being further developed into general predictors of protein-protein interactions.
